


DOI: 10.1002/aenm.201703487
一作:Chen-hao Zhang
通讯作者:Hui Xu
单位:江苏大学
全文速览:
将CO2还原为CO是合成更复杂的碳基燃料和化学品的第一步。因此,理解这一步骤对于开发用于将CO2转化为更高级产物(例如烃)的高性能电催化剂至关重要。在这里,原子铁分散在氮掺杂的石墨烯(Fe/NG)合成作为一种有效的电催化剂,用于CO2还原为CO。Fe/NG具有低的还原过电位,高法拉第效率高达80%。通过像差校正的高角度环形暗场扫描透射电子显微镜和X射线吸收精细结构分析证实了氮掺杂石墨烯层上氮限制的原子Fe部分的存在。Fe/NG催化剂为比较研究催化中心对电催化性能的影响提供了一个理想的平台。通过密度泛函理论计算进一步研究了氮掺杂石墨烯中四个氮原子包围的Fe原子(Fe-N4)的CO2还原反应机理,揭示了氮掺杂对石墨烯可能的促进作用。
背景介绍:
在使用可再生电力的燃料或化学品的电化学合成中使用CO2作为原料是一种有前途但具有挑战性的碳中和方法。理想的反应途径是将CO2直接转化为液体燃料或其他高价值的碳氢化合物。然而,这种还原过程涉及多质子耦合的电子转移步骤,对CO2的电化学还原施加高的热力学和动力学障碍。此外,只有少数材料能够将CO2还原为高级烃和含氧化合物,但具有广泛分布的C1-C3产物。另一种方法是将CO2电催化还原为CO,随后通过众所周知的费托工艺将CO转化为H2流中的烃和含氧化合物。在所有过渡金属中,已经发现银和金是用于将CO2还原成CO的最有效和选择性的电催化剂。各种地球丰富的材料和具有挑战性的碳中和方法。理想的反应途径是将CO2直接转化为液体燃料或其他高价值的碳氢化合物。然而,这种还原过程涉及多质子耦合的电子转移步骤,对CO2的电化学还原施加高的热力学和动力学障碍。此外,只有少数材料能够将CO2还原为高级烃和含氧化合物,但具有广泛分布的C1-C3产物。另一种方法是将CO2电催化还原为CO,随后通过众所周知的费托工艺将CO转化为H2流中的烃和含氧化合物。在所有过渡金属中,已经发现银和金是用于将CO2还原成CO的最有效和选择性的电催化剂。已经研究了多种地球丰富的材料和无金属的杂原子掺杂的纳米结构碳以替代基于贵金属的碳但具有挑战性的碳中和方法。理想的反应途径是将CO2直接转化为液体燃料或其他高价值的碳氢化合物。然而,这种还原过程涉及多质子耦合的电子转移步骤,对CO2的电化学还原施加高的热力学和动力学障碍。此外,只有少数材料能够将CO2还原为高级烃和含氧化合物,但具有广泛分布的C1-C3产物。另一种方法是将CO2电催化还原为CO,随后通过众所周知的费托工艺将CO转化为H2流中的烃和含氧化合物。在所有过渡金属中,已经发现银和金是用于将CO2还原成CO的最有效和选择性的电催化剂。已经研究了多种地球丰富的材料和不含金属的杂原子掺杂的纳米结构碳以替代基于贵金属的催化剂。
图文解析:
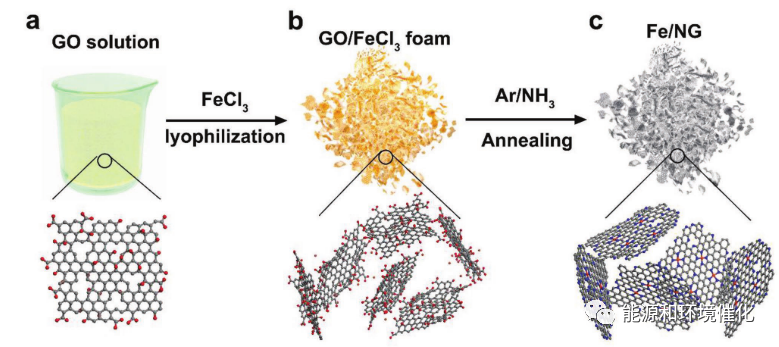
图1 Fe/NG催化剂的合成过程的示意图。a)去离子水中的GO悬浮液,B)通过冻干工艺由GO获得的Fe/NG前体泡沫,和c)通过Ar/NH3退火工艺在650-800 ℃下获得的最终Fe/NG产物。
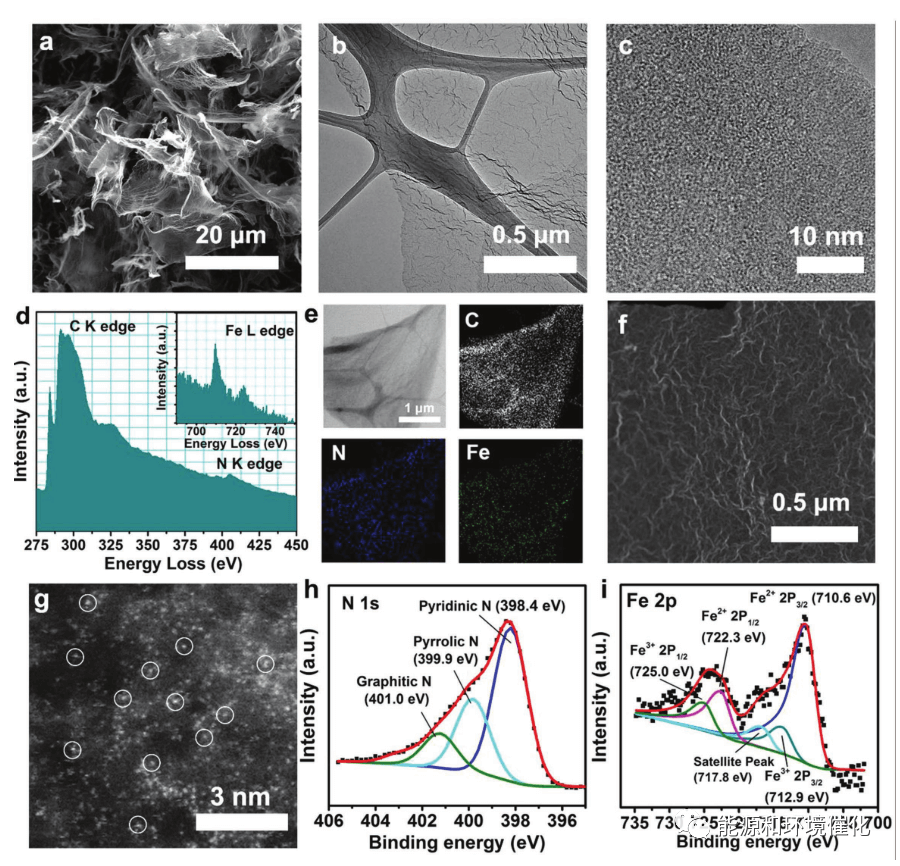
图2. Fe/NG-750的物理和形态表征。a)Fe/NG-750催化剂的SEM图像。b,c)Fe/NG-750催化剂的TEM和HRTEM图像。d)标记了Fe/NG-750的电子能量损失谱(EELS)原子谱,C K边、N K边和Fe L边(插图)。e)Fe/NG-750催化剂的STEM图像和EDS绘图。f,g)分别为Fe/NG-750催化剂的低和高放大倍率像差校正的HAADF-STEM图像。代表一些孤立的Fe原子的亮点用圆圈突出显示。h,i)分别为N 1 s和Fe 2 p3/2峰的高分辨率XPS谱。
高分辨率TEM(HRTEM)图像显示了Fe/NG-750片的薄且起皱的形态,没有形成大的金属基纳米颗粒(图2b,c)。选区电子衍射(SAED)中不存在六边形衍射图案意味着催化剂由缺陷多层石墨烯组成。然而,从电子能量损失谱(EELS)信号分析(图2d)可以明显看出石墨烯衬底上存在N和Fe物质。能量色散X射线光谱(EDS)和EELS元素映射分析进一步证实了在石墨烯片上均匀的Fe和N掺杂分布,而没有检测到Fe纳米颗粒(图1 e)。使用亚埃分辨率的像差校正HAADF-STEM技术(图2f,g);由于重元素的灵敏Z对比度,可以在石墨烯表面上清楚地观察到对应于单个Fe原子的亮点。考虑到这些亮点在石墨烯基质中的亚埃尺寸和良好的分散性,我们确定Fe物种主要作为单原子Fe位点存在。
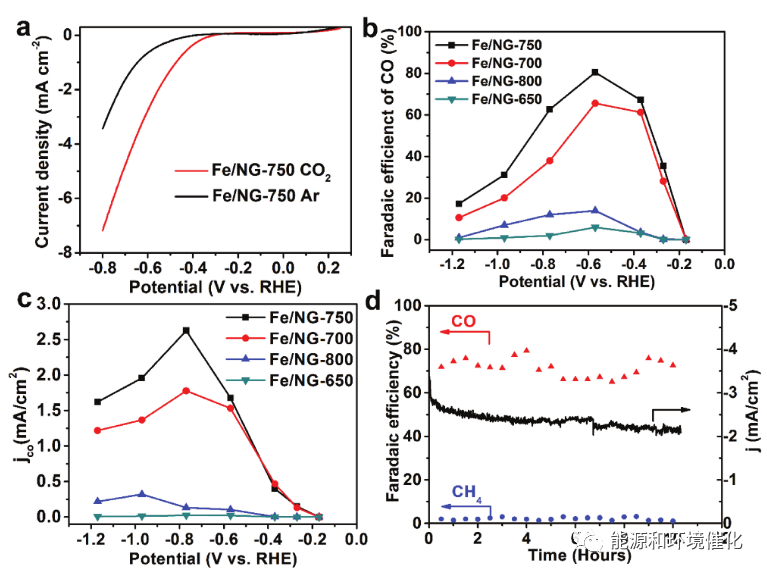
图3. Fe/NG催化剂上CO2还原为CO的电催化性能a)在Ar-或CO2-饱和的0.1 m KHCO3水溶液中,在20 mV s-1的扫描速率下,在玻璃碳电极上的Fe/NG-750催化剂的LSV曲线,催化剂负载量为0.32 mg cm-2。b)在不同退火温度下制备的Fe/NG催化剂上用于电化学CO2还原的CO的电位依赖性FE(在0.1mKHCO3水溶液中)。c)由相应的电位依赖性FE数据导出的Fe/NG催化剂上的CO的分电流密度。d)Fe/NG-750在-0.60 V下与RHE在CO2饱和的0.1 m KHCO3溶液中的稳定性试验计时电流曲线。
如图3a所示,当电位扫描到比可逆氢电极(RHE)负的-0.30 V时,还原电流急剧上升,表明Fe/NG-750催化剂上CO2还原的过电位较低。相反,在Ar饱和的0.1 m KHCO3电解质中观察到具有显著电流增加的起始电位,其源自析氢反应(HER),因为氢是检测到的唯一产物。此外,在Ar吹扫期间,在-0.80 V(vs RHE)下,CO2饱和电解质中的Fe/NG-750表现出比HER高大约两倍的电流密度。特别是对于Fe/ NG-750,在−0.60 V(相对于RHE)下,对CO的最大FE为80%(图3b)。在向Fe/NG-750催化剂施加更高的过电位的情况下,FE和CO产生的部分电流密度都逐渐下降,因为当在电极表面上检测到H2析出时,竞争性HER变得占主导地位(图2b),这也与图3a中的LSV结果一致。
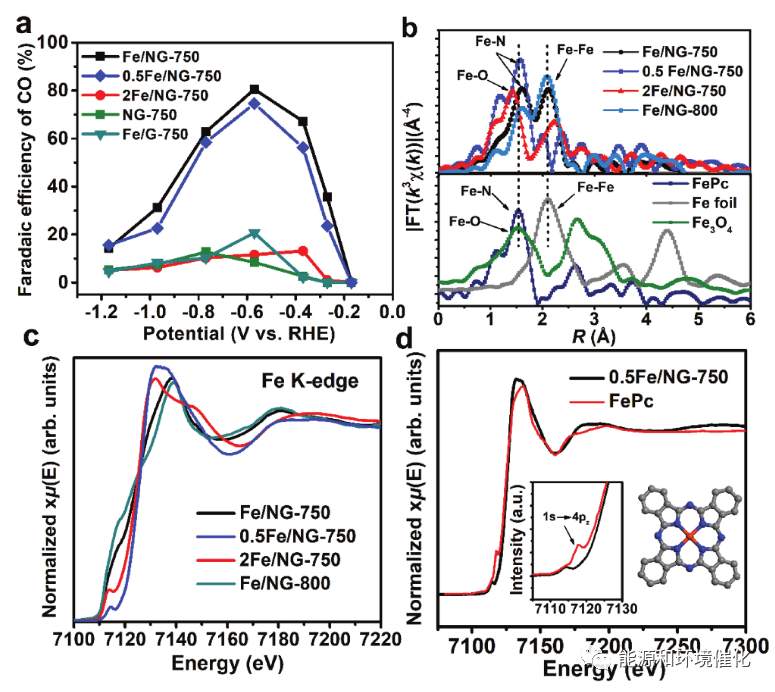
图4 催化活性和XAFS研究对Fe部分的依赖性。a)在与Fe/NG-750相比的不同条件下制备的Fe/NG对照样品的情况下,用于电化学CO2还原的CO的FE。B)Fe/NG对照样品沿着FePc、Fe箔和Fe3O4参比样品的实验Fe K边缘EXAFS光谱的傅里叶变换幅度。c)Fe/NG对照样品的归一化Fe K边缘XANES光谱。d)0.5Fe/NG-750催化剂(黑线)和FePc参比物(红线)的归一化Fe K边缘XANES光谱;插图显示边缘前特征的放大视图。
为了研究Fe结构在CO2还原活性中的作用,Fe/NG催化剂中的Fe浓度通过将添加到GO前体中的FeCl3的量加倍或减半而变化,产物分别称为2Fe/ NG-750或0.5Fe/NG-750。除了FeCl3的加入量之外,该过程与Fe/NG-750催化剂相同。HRTEM和SAED测量表明,2Fe/ NG-750催化剂由Fe 3 O 4和分散在N掺杂石墨烯表面的直径为10-20 nm的金属Fe纳米颗粒组成。如所预期的,2Fe/NG-750催化剂对CO生产表现出微不足道的选择性(FE < 10%),证实活性源自原子级Fe而不是Fe基纳米颗粒。有趣的是,0.5Fe/NG-750催化剂也显示出高的CO2对CO还原的FE(在-0.57 V下为74%),这与Fe/NG-750相当(图4a)。
总结与展望:
本研究已经证明了分散在氮掺杂石墨烯上的低含量原子Fe,用于在0.1 m KHCO3中将CO2高效还原为CO的活性。详细的结构表征,包括HRTEM,HAADF-STEM,和EDS揭示了氮掺杂的石墨烯衬底上的Fe原子的均匀分散。所制备的催化剂实现了令人印象深刻的活性,电化学还原CO2到CO在适度的过电位,沿着与高选择性和长期稳定性。Fe纳米颗粒的形成或不存在氮掺杂都降低了对照样品中的CO2还原活性,这在氮掺杂石墨烯上的Fe原子分布与电催化性能之间建立了结构-性质相关性。同步辐射表征和理论模拟表明,吡啶Fe-N4结构和氮掺杂对CO转化率的提高起着重要作用。在氮掺杂的石墨烯基质中引入微量Fe原子可以形成Fe-N4位点,用于增强CO2吸附和改善CO2活化。这项工作在均相和非均相催化剂之间建立了一座桥梁,扩大了设计和工程化石墨烯原子结构的可能性,使其具有地球丰富的金属,从而实现水溶液中高效和选择性的电化学CO2还原。
本文来自能源和环境催化,本文观点不代表石墨烯网立场,转载请联系原作者。