
缺陷对材料的功能和性能起着至关重要的作用,但通常缺乏对相关影响的理解,因为通常缺陷的低浓度使得它们难以研究。一个突出的例子是石墨烯等二维材料的拓扑缺陷。石墨烯基(光)电子器件的性能主要取决于接触电极处石墨烯/金属界面的特性。这些界面特性如何依赖于石墨烯中普遍存在的拓扑缺陷的问题具有很高的实际意义,但目前还无法回答。在这里,我们专注于典型的 Stone-Wales (S-W) 拓扑缺陷,并将理论分析与分子模型系统的实验研究相结合。我们表明,与常规石墨烯相比,嵌入的缺陷与铜表面的键合和电子转移增强。使用分子模型对这些发现进行了实验证实,其中天青芘模拟了 S-W 缺陷,而其异构体芘代表了理想的石墨烯结构。实验相互作用能、电子结构分析和吸附距离差异定量地证实了缺陷控制键合。我们的研究揭示了缺陷对石墨烯/金属界面电子耦合的重要作用,并表明拓扑缺陷工程可用于性能控制。
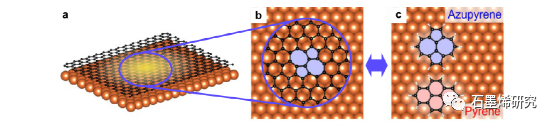

图 1. 根据理想石墨烯晶格和嵌入缺陷的拓扑结构选择分子模型系统 (a)。具有拓扑缺陷的石墨烯/金属界面(b,c)。拓扑 S-W 石墨烯缺陷与金属表面 (b) 的局部相互作用通过应用于嵌入缺陷和相关分子模型系统 (c) 的计算和实验方法的组合来研究。这些模型系统提供了对嵌入缺陷无法获得的信息的访问。
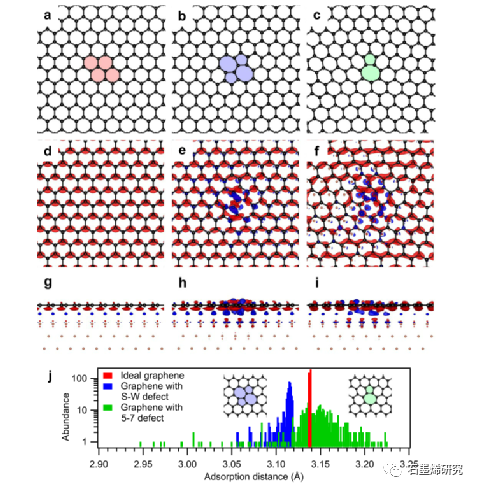
图 2. 拓扑石墨烯缺陷与 Cu(111) 表面的相互作用。左:理想的石墨烯晶格,中:嵌入 S-W 缺陷,右:嵌入 5-7 缺陷。(a-c)自支撑石墨烯晶格的优化结构。(e,f)吸附在Cu(111)上的三种石墨烯结构的电荷密度差异图的俯视图和(g-i)侧视图;等值面值:0.0005 e-/Å3,蓝色:电子积累,红色:电子耗尽。( j )Cu(111)基板上方碳原子吸附距离的丰度分布。红色:理想石墨烯,蓝色:具有 S-W 缺陷的石墨烯,绿色:具有 5-7 缺陷的石墨烯。选择具有 456 个碳原子的大晶胞以减少相邻缺陷之间的横向相互作用。
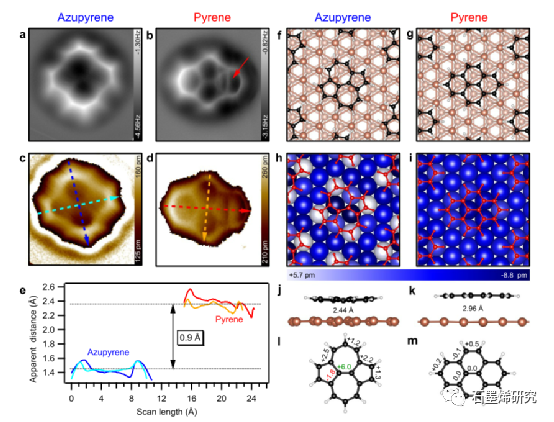
图 3. LT-AFM 测量揭示了模型缺陷 (Azupyrene) 和参考 (Pyrene) 在 Cu(111) 上的化学结构和表观吸附距离,与 DFT 计算一致。(a,b)z = -100 pm(a)和 z = -50 pm(b)相对于 U = 100 mV 和 I = 20 帕。明亮的垂直线(红色箭头)和芘分子右手环的伸长是由于尖端诱导的分子运动而产生的图像伪影。(c,d)从 Δf ( z )光谱的三维网格测量获得的Pyrene和Azupyrene的重建形貌图像(即吸附距离)。(e)Azupyrene(蓝色,青色)和Pyrene(红色,黄色)的形貌扫描线显示分子表观吸附距离的差异。沿着(c,d)中的虚线箭头获取扫描线。(f,g)DFT 优化吸附结构的俯视图。(h,i)与松弛的清洁表面相比,最上面的铜原子的垂直位移(以 pm 为单位)。正值(较浅的蓝色阴影)表示向分子的位移。( j , k )具有计算的垂直粘合距离的侧视图。(l,m)相互作用引起的碳 – 碳键长度相对于气相结构的变化(以 pm 为单位)。
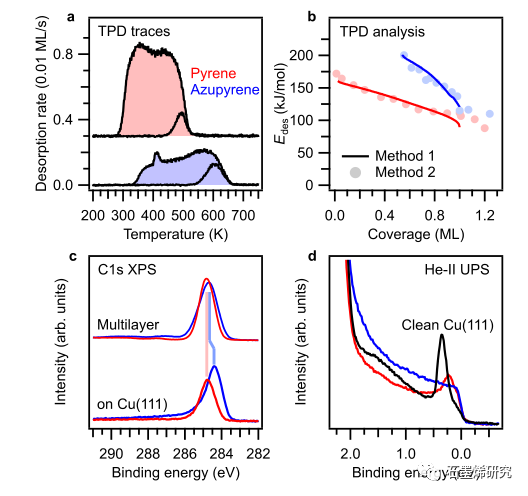
图4。TPD和光电发射(XPS,UPS)数据表明模型缺陷和金属之间的结合增强。(a)初始单层覆盖和较低初始覆盖的TPD痕量的Azupyrene和Pyrene。分析中使用的中间覆盖层的TPD数据见补充图S6。(b)通过使用两种不同方法分析TPD数据获得的覆盖范围相关解吸能量。(C)Cu(111)上多层膜和单层膜的C 1s光电子能谱。两者之间的相对位移用阴影线标出。Azupyrene单层峰的不对称性表明与表面金属态的杂化。(d)清洁表面(黑色)和芘和芘的单层的He-II UP光谱。表面态的消失(清洁表面上的结合能为0.3 eV)显示了对氮杂芘的更强的相互作用。
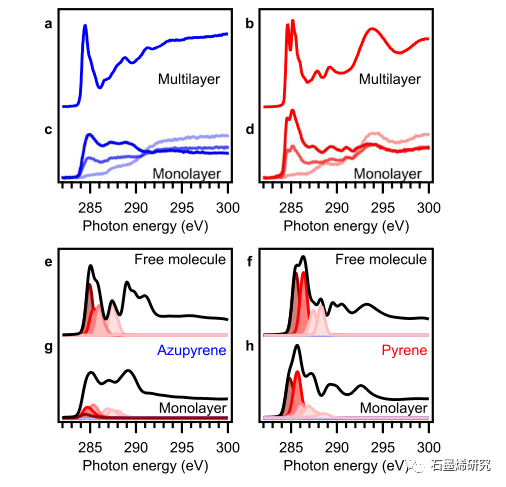
图 5. 碳 K 边 NEXAFS 数据揭示了模型缺陷Azupyrene未占轨道的相互作用引起的变化。实验光谱:(a,b)多层;(c,d)(a,c)Azupyrene和(b,d)Pyrene的单层。多层光谱是用相对于表面法线方向为 53° 的电场矢量拍摄的,而单层光谱是用配色方案指示的角度拍摄的(25°,粗体色;53°,中间色;90°,微弱 颜色)。自由分子 (e,f) 和单分子层 (g,h) 的 DFT 计算的 NEXAFS 光谱的 MO 投影分析:(e,g) Azupyrene; (f,h) 芘。LUMO 的贡献为深红色,更高轨道的贡献为逐渐变浅的颜色;黑色的总光谱。计算的光谱被严格地移动了 -6 eV 以匹配实验能量标度。
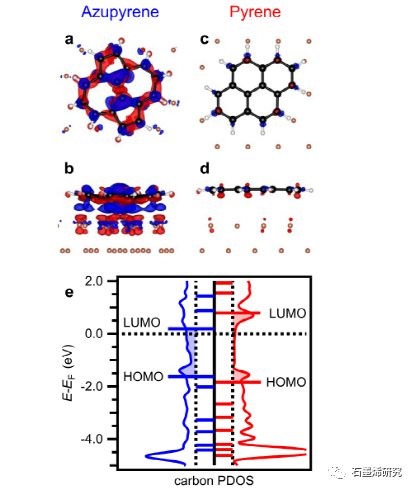
图6. 基于DFT的成键分析显示电子从Cu(111)表面转移到未被占据的Azupyrene的LUMO中,并且芘没有电荷转移。(a d)氮杂芘(a,b)和芘(c,d)的电荷密度差异图。所有图的等值面值都是0.002e−/Å3。红色:电子耗尽,蓝色:电子积累。(Azupyrene(左)和Pyrene(右)的碳投影DOS,水平线表示气相轨道能量,移动以说明功函数。子图(a,b)显示了与S-W缺陷周围电子重排的相似性(见图2e,h)。
相关研究成果由吉森大学André Schirmeisen和马尔堡大学J. Michael Gottfried等人2022年发表在ACS Nano (https://doi.org/10.1021/acsnano.2c01952)上。原文:Topological Stone–Wales Defects Enhance Bonding and Electronic Coupling at the Graphene/Metal Interface。
本文来自石墨烯研究,本文观点不代表石墨烯网立场,转载请联系原作者。