
研究背景
氧化石墨烯作为倍受关注的二维材料,仅有一个碳原子厚度,其片层表面上主要分布环氧和羟基官能团而片层边缘分布着羧基官能团。氧化石墨烯在分子监测传感器、生物医学和药物传递、海水淡化及环境水处理、新材料新能源等等诸多领域具有非凡应用前景,然而,氧化石墨烯分子结构特征及属性的基础理解却一直是未解的难题。
许多不同模型尝试描述氧化石墨烯的分子结构,Lerf-Klinowski模型是目前最广泛使用的,认为氧化官能团随机分布在氧化石墨烯片层表面。扬州大学涂育松课题组与上海大学石国升课题组,于2014年合作运用DFT量子计算分析Hummers制备法生产氧化石墨烯中的反应过程,获得高锰酸根氧化石墨烯的反应动力学过程,并由此提出基于DFT计算的全新石墨烯结构模型(Shi-Tu模型),展示首个基于DFT计算的氧化官能团分布结构 [Angew. Chem. Int. Ed. 53, 10190 (2014)]。Shi-Tu模型表明,氧化石墨烯表面的氧化位点具有高度相关特性,并表现为大面积亲水区域(含氧化官能团)和疏水区域(不含氧化官能团)的共存特征,从而证实了近年来表征技术,如超高分辨率TEM或固体NMR,对氧化石墨烯结构的实验观察。
内容简介
近日,扬州大学涂育松课题组和上海大学石国升课题组再次合作,针对氧化石墨烯结构提出全新认识。首次实验和理论证实:在常温下,水分子吸附导致氧化石墨烯转换成自发动态共价材料,其表面氧化官能团的C-O化学键自发断开或重新形成,从而实现沿着氧化石墨烯表面的自发的氧动态迁移运动。对照氮干环境和加水条件,同步辐射原位红外傅立叶光谱实验,清楚显示了氧化石墨烯表面环氧和羟基的化学含量实时自发的类周期性的振荡变化,二维显微红外成像进一步给出了这些氧化官能团的自发动态二维分布。更为重要的是,当氧化石墨烯吸附了生物分子(如:胞嘧啶)后,原本动态分布的氧化官能团重新恢复稳定分布,此表明了氧化石墨烯上的氧化官能团与生物分子之间存在自适应性的相互作用。DFT量子计算和从头算量子动力学模拟证实氧化石墨烯上氧化官能团迁移的反应路径,发现正是由于水分子吸附极大降低了环氧和羟基中C-O键断裂/形成的可逆反应能垒,使其低于或相当于液态水中氢键能量,从而带来了氧化石墨烯的自发动态共价材料特征。
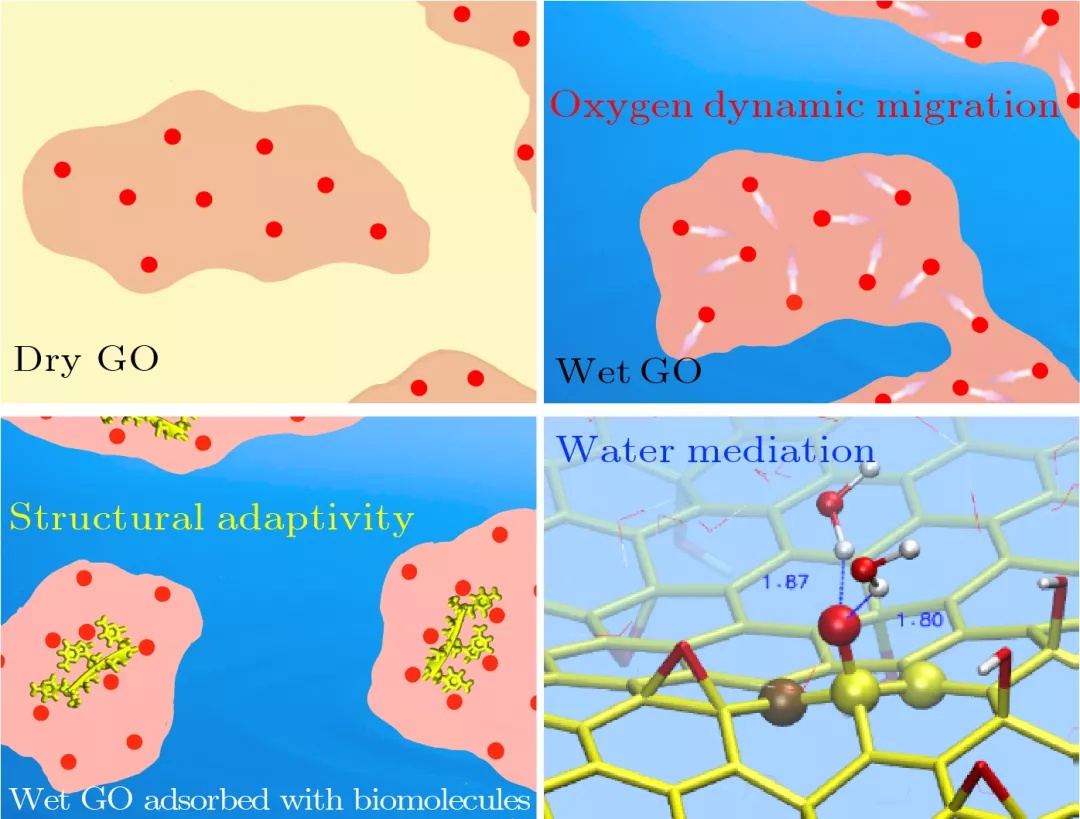

图:氧化石墨烯常温下转换成的自发动态共价材料示意图及分子机制。左上:在干燥条件下氧化石墨烯上的氧化官能团静态分布示意图,亮红色区域代表氧化石墨烯的亲水区(含氧化官能团的氧化区,红圆点代表氧化官能团),其他区域为疏水区(不含氧化官能团的非氧化区);右上:原位吸附水分子后湿氧化石墨烯上的氧化官能团动态迁移运动及其导致的氧化官能团动态分布,箭头表示氧化官能团的移动性;左下:原位吸附生物分子后氧化石墨烯上氧化官能团自适应性分布,黄色线分子为吸附上的生物分子;右下:氧化石墨烯原子结构中,水分子诱导下氧化官能团的动态迁移过程,碳显示为黄色,氧为红色,与迁移相关原子显示为小球。
通常,氧化石墨烯的精细结构TEM或NMR直接观察要求干燥甚或真空条件,之前研究中氧化石墨烯的动态特性从未被发现。事实上,氧化石墨烯上的氧动态迁移也是首先在理论计算中被预测,然后再被原位红外光谱实验证实;氧化石墨烯通常会以多个片层堆叠出现,理论计算发现片层之间氧迁移需要跨越很高能垒,同时水分子吸附导致层间距增加而层间迁移能垒会进一步增加,这表明实验上所观察到的氧化官能团动态迁移主要为沿着单片氧化石墨烯表面的动态迁移行为。
实现动态共价键分子界面需要设计各种不同可逆化学反应,但通常这些反应要求一定催化或温度或pH等条件。氧化石墨烯界面是首次报道的在常温下可实现的大面积的自发动态共价分子界面。审稿人评论,此研究结果增加了对氧化石墨烯的基本理解,挑战了广为接受的“静态图片”(即,氧化石墨烯在其界面动力学过程中纯粹表现为一个被动角色),同时氧化石墨烯界面氧的动态移动性的观察,对于大量的具有类似特征的其他系统是非常重要的。
考虑到实际环境(如生物环境)中,氧化石墨烯通常会不可避免地接触到气态或液态的水,氧化石墨烯的自发动态共价特性以及分子吸附响应的自适应性相互作用,提供了氧化石墨烯应用中关键的基础理解和认识,开辟了一条设计自适应动态响应系统的新途径。
本文来自ChinesePhysicsL的个人博客,本文观点不代表石墨烯网立场,转载请联系原作者。