
背景
二氧化碳的捕获和储存(CCS) 是当前环境保护和可持续发展中的一个热点问题,通过降低大气中的二氧化碳浓度来适应气候变化已经得到了广泛的研究和开发。
二氧化碳捕获有不同的分离技术:a)吸附,b)吸收,c)低温蒸馏,d)膜分离,e)气体水合物,f) 化学链。
除低温分离外,其余的方法都需要一些材料作为载体。具有相似直径和形状的气体分子的分离是一个重要的研究领域。
膜材料分离技术是根据不同气体和膜材料之间的物理和化学相互作用的差异来分离气体。这将允许一些成分根据尺寸(动力学)和亲和力(热力学)优先通过膜。膜技术可以通过减小设备尺寸和降低能源需求而成为CO2捕集过程的有力工具。
主要内容
目前的研究涉及不同功能原子(C和H)和不同孔径的孔石墨烯对CO2/H2气体混合物的渗透率和选择性。我们采用反应分子动力学模拟(ReaxFF MD)和密度泛函理论(DFT)计算,以便深入了解通过孔隙的分子的渗透和相互作用特性。
其中相互作用能和能垒计算公式如下:
Eint=EM/Graph – EGraph -EM
Ebarr=Eint.max – Eint.min
在本工作中,模拟系统尺寸为18 Å×22 Å×120 Å。气体盒中包含中有50个CO2和50个H2分子,在气体混合物的两侧放置两个石墨烯片,以提高纳米孔膜的透气性。并且石墨烯膜的两侧之外还有真空空间。
为了避免石墨烯层由于气体分子施加的压力而发生位移,在模拟过程中固定了石墨烯中两个碳原子的位置。所选择的原子位于尽可能远离孔隙的地方,以模拟边缘附近原子的真实行为。
最后进行了300 K下的NVT系综模拟,时间步长为0.5 fs。vdW相互作用的截止距离为12 Å。采用Ewald描述静电相互作用。对每个系统进行4 ns 动力学模拟。
结果与讨论
气体分子与石墨烯表面的相互作用
首先利用ReaxFF力场计算H2、CO2和石墨烯体系的结构性质,并将结果与DFT-B3LYP方法进行比较。结果表明两种计算结果吻合较好。
其中得到的结构参数列于表所示。计算得到的结构参数也与实验值吻合较好。
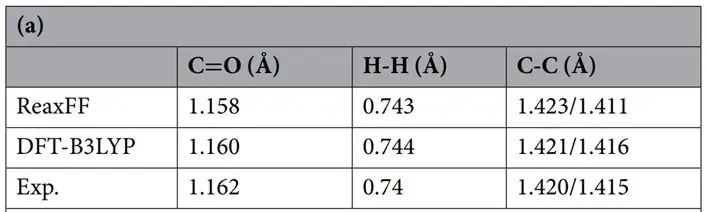

H2和CO2分子被放置在石墨烯薄片表面,其分子轴与薄片平行。采用ReaxFF和DFT-B3LYP两种计算方法对研究系统进行了全面的结构优化。用ReaxFF对整个体系进行充分的结构优化后,我们发现H2和CO2分子分别以约3和3.3 Å的平衡距离漂浮在石墨烯表面(见图a)。对比B3LYP-D3/TZVP计算结果,结果表明ReaxFF与DFT-B3LYP结果吻合较好。
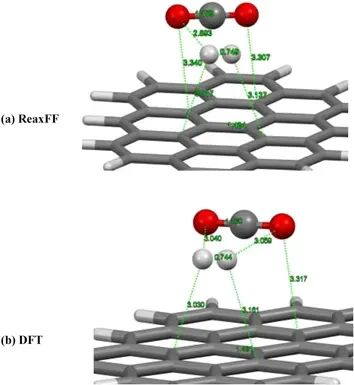
两种理论方法计算出的吸附H2和CO2分子的键长值几乎相同。ReaxFF与DFT-B3LYP方法结果的一致性足以说明ReaxFF力场适用性。
气体分离的MD模拟
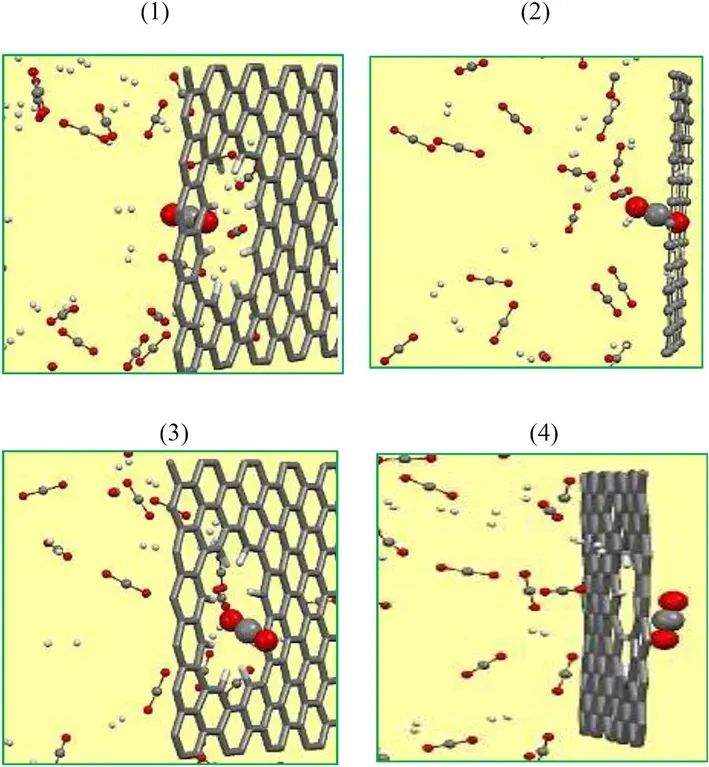
设计了一些双层多孔石墨烯模型,这些模型具有不同尺寸和功能原子,彼此之间的距离为30 Å。多孔石墨烯模型的示意图如图所示。从图中可以看出,通过从石墨烯晶格中去除碳原子获得了一系列孔径。根据CH原子数量来命名多孔石墨烯模型,即10CH,12CH等。
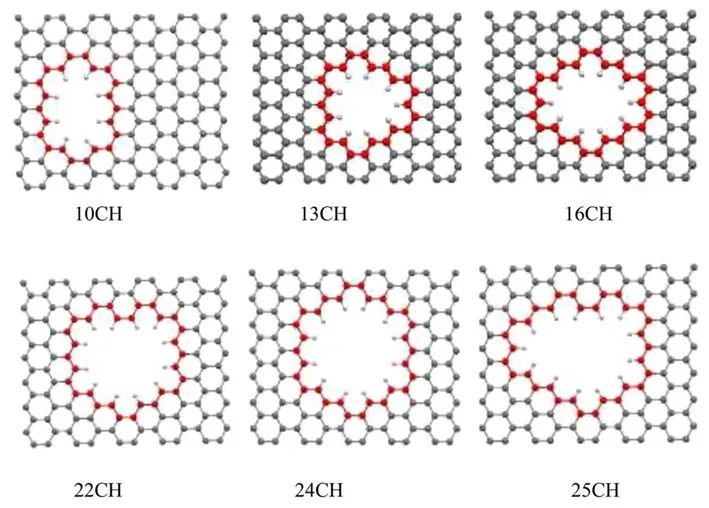
为了研究孔的大小和形状对CO2/H2分离的影响,确定什么样的孔在选择性和渗透性之间具有良好的平衡是很重要的。
渗透性是特定气体通过膜的通量,而选择性(渗透比)是两种气体分子渗透事件的次数之比。可以推断,当渗透比等于1时,不存在选择性,渗透比越大,选择性越高。
孔径定义为孔内最短和最大距离的平均值,孔面积根据删除的苯环面积计算得到。其中膜的渗透率:F=N(mol)/S(m2).t(s)
其中N为两个方向通过膜的气体分子的摩尔数,S为膜的总面积,t为持续时间。在4 ns MD模拟后,估计通过膜的分子数量,并将其列在下表中。
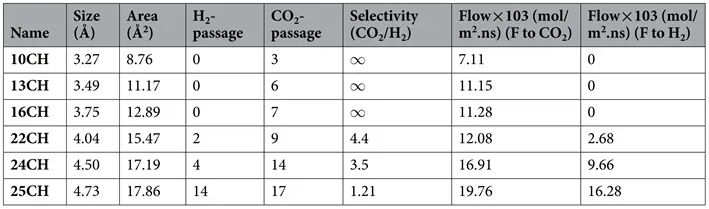
10CH膜对CO2/H2混合物具有良好的CO2分子选择性。随着孔径的增大(24CH和25CH孔径分别为4.50和4.73 Å),选择性越低,渗透率越高。这是因为孔的大小足以让CO2和H2分子渗透。孔径为3.27 Å的10CH具有较高的选择性,但其渗透性较差,不利于CO2/H2的分离。综上,孔径为3.75 Å的16CH孔具有较高的选择性,是CO2/H2分离的最佳候选孔。
进一步评估了最佳16CH膜的作用效果,延长模拟时间,发现即使在10 ns后也没有H2通过孔。下图d为模拟时间10ns后含有CO2/H2混合物的16CH膜的动力学快照。
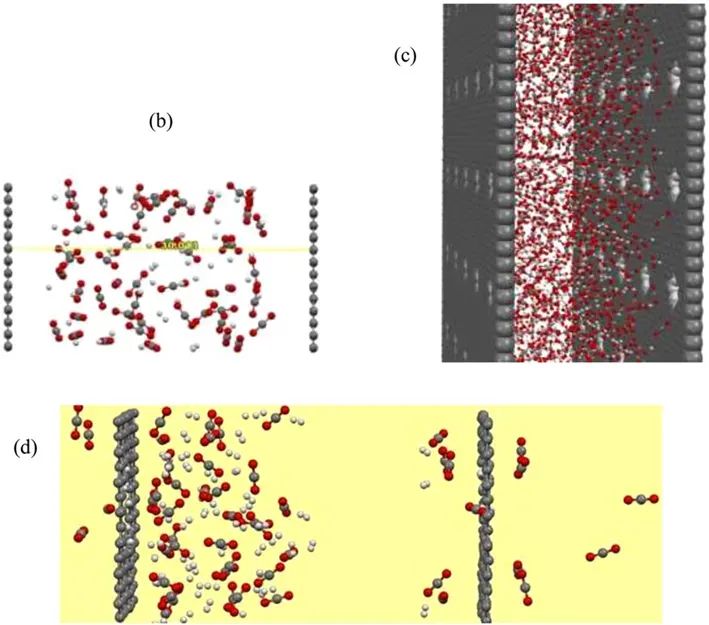
这种优越的选择性可能是由于孔边缘正电荷(H原子)和CO2分子边缘的O原子之间的吸引力导致CO2向孔迁移,而这些正电荷将H2分子从孔边缘排斥。
CO2气体分子穿透16CH孔的机理
根据MD模拟观测,可以将渗透过程如下所述:首先分子靠近膜孔移动,然后到达膜的另一边。在模拟过程中,CO2和H2分子都接近石墨烯表面。随后一个CO2分子在孔隙周围来回移动,最终克服势垒能,43 ps时垂直穿过孔隙。CO2分子通过16CH孔的过程快照如图所示。之后,越来越多的CO2分子通过孔,而经过10ns的模拟时间后,仍然没有H2跃迁,这表明16h孔对CO2/H2分离具有很高的选择性。
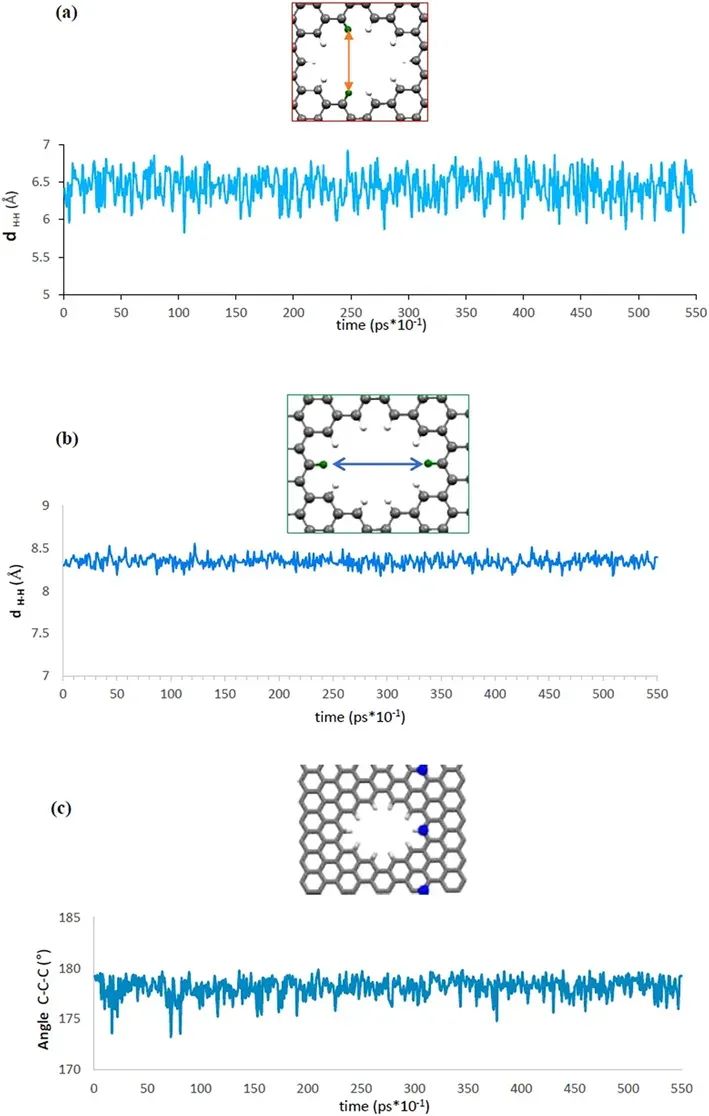
孔结构的状态
计算了孔结构中两个水平H原子距离、两个垂直H原子距离以及三个C原子的夹角。
比较两个H原子之间的平衡距离可以看出,垂直方向的H原子比水平方向的H原子波动更大。这一发现表明,垂直H原子在气体通过孔的过程中起关键作用。此外,石墨烯片偏离平面,其表面角度在5°左右波动。
气体穿透过程中的电荷
为了进一步了解气体渗透过程中与孔结构的相互作用,本部分专门研究了通过孔的分子与边缘相邻原子之间的电荷转移。由于在本工作中使用了ReaxFF力场,所以可以评估在渗透过程中转移的电荷量。采用QEq方法分析了CO2分子在1 ps (CO2分子离孔较远)和10 ps (CO2分子被困在孔内)时通过孔的CO2分子与孔内H原子之间的电荷。图表示所选原子上的电荷量。
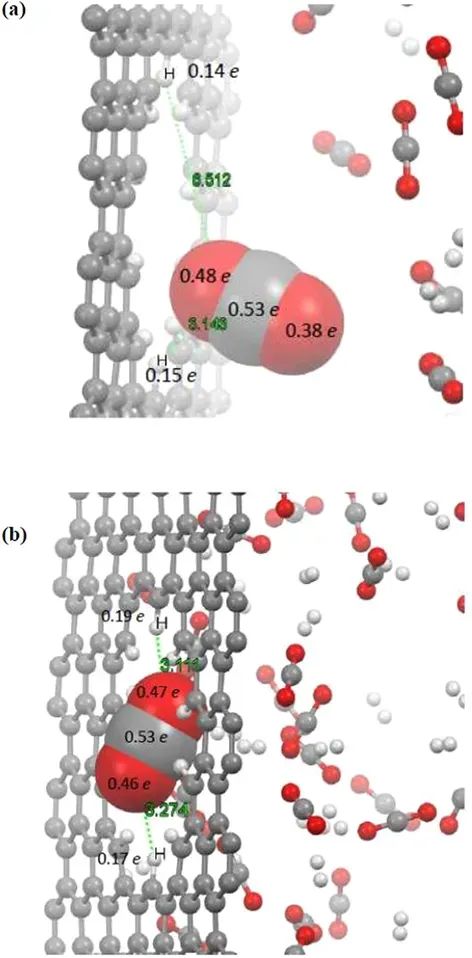
当CO2的O原子位于孔隙的H原子之间时,O原子上的电荷增强。在渗透过程中电荷已经从边缘的H原子转移到孔中捕获的CO2分子的O原子上。
气体分子穿过孔的垒能
考虑了H2/CO2气体分子通过孔中心的两种构型;即接近孔中心的分子轴的平行方向和垂直方向。利用DFT-D与ReaxFF的相互作用结果进行比较(c:DFT-D结果;d:ReaxFF结果)。
计算得到的能垒如表所示。
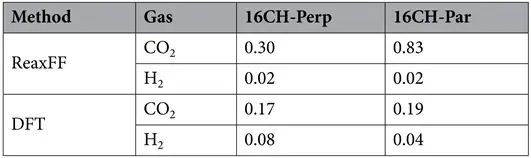
DFT计算结果表明,CO2分子倾向于以分子轴垂直于石墨烯表面的方式通过孔道。数值大小趋势上,ReaxFF计算结果与DFT计算结果定性一致。从上图气体分子与孔之间的相互作用曲线可以看出,穿孔的过程中,气体分子与孔的相互作用先变强后变弱,气体分子在孔的中间位置时相互作用最强。
从表中的能垒数据得到H2分子通过16CH孔的势能小于CO2分子,这表明氢分子更容易穿过膜。然而,从MD模拟结果中我们可以发现,CO2分子通过孔,而H2分子被拒绝!!!
这种差异可以用这样一个事实来解释,即相互作用能在气体分子通过孔的渗透中起重要作用。从孔与气体分子的相互作用能可以看出,CO2分子的相互作用强度比H2分子的要大,导致CO2分子被吸引到孔边缘,然后来自其他CO2气体的吸引的压力推动,孔边缘的CO2很容易穿透孔。同时,CO2具有显著的四极矩,增强了氢修饰石墨烯的色散吸引力。这证实了MD模拟的结果,即只有CO2分子可以通过16CH膜的孔。从而表明16CH对CO2/H2分离具有高选择性。
结论
通过对石墨烯结构的改变,16CH膜材料显著提高了膜对CO2/H2分离的选择性和渗透性。
CO2气体在渗透过程中,电荷从孔中的H原子转移到被捕获CO2分子中的O原子上。这表明通过的CO2分子与孔膜之间的相互作用相当强。
通过气体穿越能垒表明,H2分子穿孔能垒更低,但MD 结果只有CO2分子穿过孔结构,主要因为孔结构和CO2之间具有更强的相互作用;其他气体分子的压力作为驱动力,推动孔边缘的分子穿过孔结构。
本结果从原子、电子角度揭示了气体渗透的机理,为构建具有创新分离性能的新材料提供了一条很有前景的途径。
文献信息:DOI:10.1038/s41598-017-14297-w
本文来自Materials Studio,本文观点不代表石墨烯网立场,转载请联系原作者。